• 3 min de lectura
El diseño de baterías recibe un impulso gracias al aprendizaje automático a escala atómica
Investigadores del LLNL utilizaron aprendizaje automático informado por la física para modelar ánodos de iones de sodio y electrolitos de iones de litio a escala atómica.
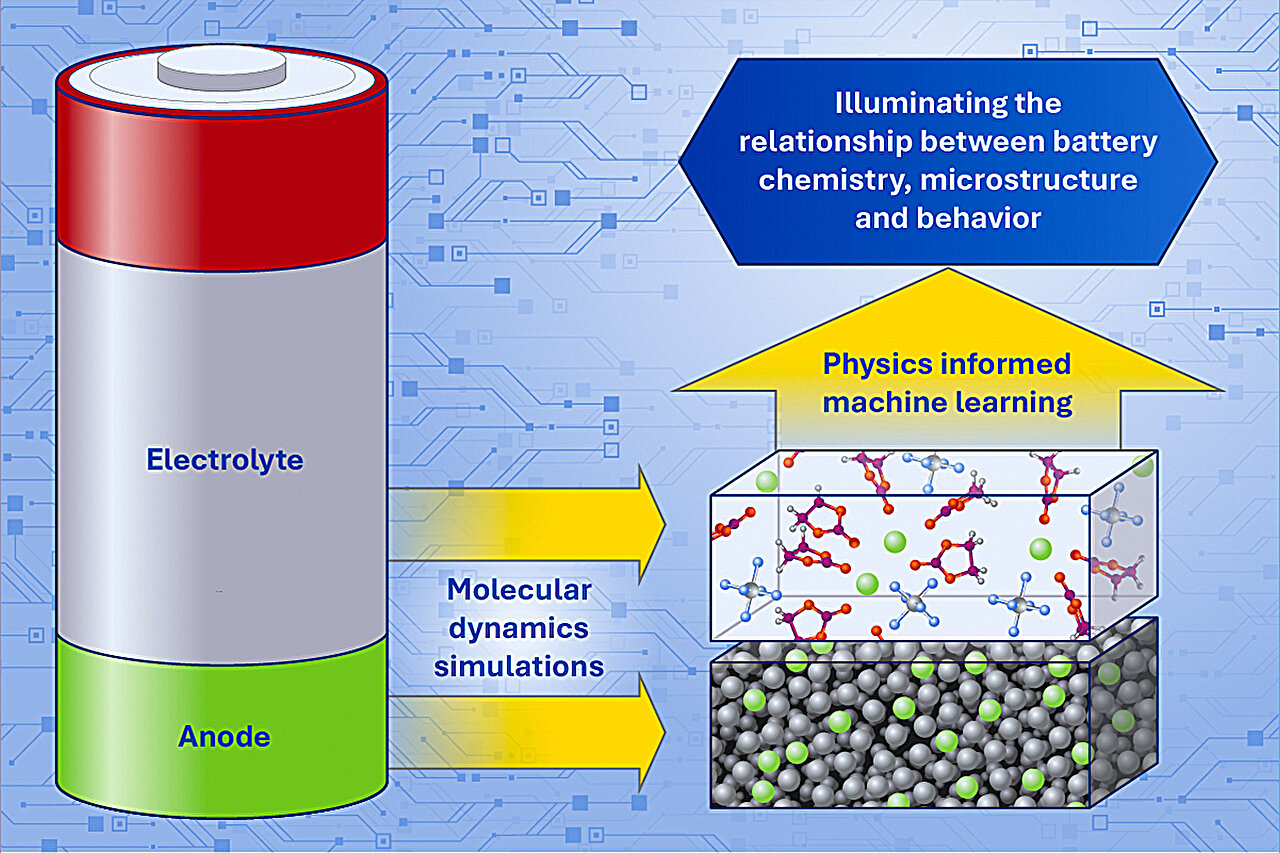
Imagen: TechXplore
Investigadores del Lawrence Livermore National Laboratory (LLNL) afirman que el aprendizaje automático informado por la física puede convertir uno de los mayores problemas de la ciencia de baterías —el desorden a escala atómica— en una herramienta de diseño.
En dos artículos de 2026, el equipo combinó simulaciones de dinámica molecular con aprendizaje automático para estudiar ánodos de carbono duro para baterías de iones de sodio y electrolitos líquidos para baterías de iones de litio. Según la científica del LLNL Liwen (Sabrina) Wan, el trabajo muestra que la complejidad estructural no es solo una barrera para la comprensión sino potencialmente una ventaja para diseñar materiales de almacenamiento de energía de próxima generación.
Carbono duro en baterías de iones de sodio
El primer estudio, publicado en Energy Storage Materials, se centró en el carbono duro, el material de ánodo con mayor madurez comercial para las baterías de iones de sodio. La abundancia del sodio y su disponibilidad nacional hacen que esta química sea atractiva para la resiliencia de la cadena de suministro en EE. UU., pero el carbono duro es difícil de diseñar porque consiste en láminas desordenadas, parecidas al grafeno, llenas de poros y vacíos.
El LLNL usó sus sistemas de computación de alto rendimiento para simular cómo se mueven los átomos en el material con el tiempo. Eso produjo lo que el autor Nikhil Rampal describió como una película, átomo por átomo, de iones de sodio difundiéndose, agrupándose o quedando atrapados dentro del carbono. El equipo luego entrenó un modelo de aprendizaje automático con esas simulaciones, lo que permitió ejecuciones más grandes, más largas y más precisas a menor costo.

Recomendado
Los satélites Luch-5 mantuvieron a la Soyuz MS-29 conectada a la ISS
El modelo clasificó el movimiento de los iones de sodio en ocho regímenes diferentes vinculados a las interacciones de los iones con el carbono duro. Los investigadores encontraron que, a medida que aumentan la densidad del carbono y la carga de sodio, los iones pueden agruparse o quedar atrapados en nanoporos, lo que afecta la capacidad a altas tasas y la seguridad térmica. LLNL afirma que el resultado es un mapa cuantitativo que vincula la microestructura con el transporte iónico, con orientación práctica para mejorar el rendimiento del carbono duro.
Cribado de electrolitos para baterías de iones de litio
El segundo artículo, publicado en EES Batteries, aplicó el mismo enfoque a los electrolitos de baterías de iones de litio. El diseño de electrolitos es notoriamente difícil porque las combinaciones de disolventes, sales, aditivos y concentraciones son demasiado numerosas para probarlas exhaustivamente.
En lugar de confiar en representaciones moleculares basadas en texto, el equipo del LLNL generó configuraciones moleculares tridimensionales realistas mediante dinámica molecular y las introdujo en un modelo de aprendizaje automático que predijo la estabilidad estadística de cada configuración. Los investigadores sostienen que la estabilidad electroquímica depende del conjunto molecular completo, no solo de una lista de ingredientes.
Rampal dijo que cambiar una sal de litio produjo una ventana de estabilidad un 57 % más amplia, impulsada por cómo el anión se organizaba alrededor del ion litio —un efecto que los codificadores convencionales basados en texto pasarían por alto.
Wan dijo que el flujo de trabajo más amplio podría convertirse en una plataforma de cribado de alto rendimiento para materiales de baterías en químicas de litio, sodio y multivalentes.
Los dos artículos son:
- Nikhil Rampal et al, Physics-informed machine learning exploration of Na storage mechanisms in disordered carbon, Energy Storage Materials (2026), DOI: 10.1016/j.ensm.2026.104967
- Srikant Sagireddy et al, Integrated machine learning-molecular dynamics framework for electrolyte property prediction, EES Batteries (2026), DOI: 10.1039/d6eb00024j

Frontier Editor
Dan is our resident futurist, covering electric mobility, space exploration, and the smart home. He's interested in atoms just as much as bits. Whether it's a new battery chemistry, a reusable rocket, or a protocol that finally makes IoT devices talk to each other, Dan breaks down the engineering that pushes humanity forward.
vía TechXplore


